Introduction
Despite decades of research, neurodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s disease (PD) remain among the most formidable challenges in medicine. Recent approvals of anti-Aβ antibodies have demonstrated, at least modestly, that removal of pathogenic protein aggregates can translate into clinical benefit for AD patients. These approvals have energized the field, fueling next-generation efforts to produce more brain-penetrant anti-Aβ antibodies and to extend the similar approach to other protein aggregates, including α-synuclein.
Yet antibody-based approaches face intrinsic limitations. Antibodies are largely restricted to extracellular targets, and thus cannot directly clear intracellular aggregates such as phosphorylated tau tangles or α-synuclein inclusions. Even when they succeed in aggregate removal, they do not address the upstream deficit that drives aggregate formation in the first place—the age-related decline of the autophagy–lysosomal pathway (ALP).
As neurons age, their capacity to recycle proteins and organelles diminishes, leading to accumulation of toxic substrates and progressive dysfunction. A more durable therapeutic strategy may lie in restoring the cell’s intrinsic housekeeping capacity—rejuvenating the ALP so neurons once again clear aggregates “from within,” much as they do in youth. This vision naturally leads to TRPML1, a lysosomal ion channel that sits at the center of lysosomal homeostasis and cellular clearance.
Genetic Linkage Between TRPML1 and Neurodegenerative Diseases
A common misconception is that TRPML1 is not genetically linked to age-related neurodegenerative diseases such as AD and PD, since most GWAS studies have not flagged it among the many ALP-related risk genes. This absence, however, reflects the rarity and nature of TRPML1 mutations rather than their biological relevance.
Unlike more frequently altered genes such as GBA1, pathogenic TRPML1 variants are extremely rare. Over 95% are frameshifts or deletions that abolish channel function, producing mucolipidosis type IV (MLIV)—a severe lysosomal storage disorder characterized by neurodevelopmental deficits and progressive neurodegeneration. These mutations provide a natural experiment: complete loss of TRPML1 function devastates lysosomal biology and the nervous system.
Even subtle disruptions reaffirm its role in neurodegeneration:
- Single-point mutations that reduce TRPML1 activity cause severe oromandibular dystonia and early-onset parkinsonism (Ghasemi et al., CJNS, 2025).
- Heterozygous variants are linked to Lewy Body Disease (LBD) and Alzheimer’s disease with Lewy Body Variant (ADLBV) (Clark et al., PLOS One, 2015).
Taken together, these insights establish TRPML1 as a genetically validated target. If complete loss leads to profound neurodegeneration and partial loss predisposes to PD- and AD-related phenotypes, then carefully restoring or enhancing its activity represents a rational therapeutic strategy. In fact, TRPML1 may be underappreciated by conventional GWAS precisely because of its rarity—yet as a master regulator of the autophagy–lysosomal pathway, it has the potential to deliver broader therapeutic benefit than interventions aimed at single risk genes.
This unique genetic profile not only underscores TRPML1’s causal role in neurodegeneration but also strengthens the case for therapeutic agonism—providing a foundation for why TRPML1 is such a compelling target for disease modification.
Why TRPML1 Is a Compelling Target
Building on its genetic validation, TRPML1 is far more than a regular risk gene — it functions as a master regulator of the autophagy–lysosomal pathway (ALP). Its activation initiates a cascade of protective processes, including lysosomal calcium release, TFEB/TFE3 activation, lysosome biogenesis, autophagosome formation, enhanced autophagic flux, and lysosomal exocytosis (Figure 1). In effect, TRPML1 sits at the top of the regulatory hierarchy that sustains cellular proteostasis and resilience.
This systemic control is what makes TRPML1 uniquely powerful. The pathology of AD, PD, and other neurodegenerative diseases arises not from the dysfunction of a single protein, but from the global decline in ALP activity with aging. Unlike antibody-based therapies that can only clear specific extracellular aggregates such as Aβ plaques—and are far less effective against intracellular lesions like pTau tangles or α-synuclein inclusions—TRPML1 agonism addresses the root cause: impaired cellular clearance capacity.
By reactivating the ALP at multiple levels simultaneously, TRPML1 agonists have the potential to restore homeostasis more effectively than interventions aimed at single downstream proteins. This systems-level approach is what distinguishes TRPML1 from nearly all other neurodegeneration targets currently under development.
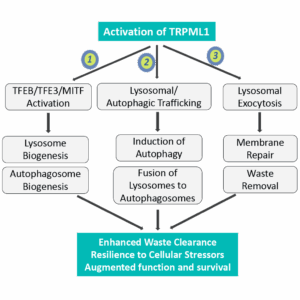 Figure 1.
Figure 1.
TRPML1 serves as a central switch for the autophagy–lysosomal pathway, coordinating multiple downstream mechanisms that restore proteostasis and resilience in aging neurons.
Challenges in Developing a Pharmaceutically Viable TRPML1 Agonist
Since the discovery of TRPML1 as an endolysosomal ion channel (Dong et al., Nature, 2008), many groups have attempted to develop agonists, but most efforts have stumbled on what one competitor aptly called a “physicochemical nightmare.” The root challenge lies in the binding pocket: deeply buried, highly hydrophobic, and shielded within the lipid bilayer (Figure 2).
As a result, potent agonists are often themselves highly hydrophobic, leading to poor solubility, metabolic instability, and limited bioavailability. Attempts to introduce polarity can improve solubility and oral exposure, but usually at the cost of brain penetration — which is non-negotiable for both efficacy in the target organ and maintaining an adequate therapeutic margin over systemic exposure.
Encouragingly, recent advances in structure-based drug design (SBDD) have shown that these trade-offs can be balanced. By carefully tuning non-polar interactions and metabolic stability, we have generated TRPML1 agonists that are highly brain-penetrant, orally bioavailable, and potent. One such molecule has now advanced through IND-enabling studies with a strong safety profile, positioning it to enter clinical testing. What once seemed an insurmountable barrier has become a tractable path forward.
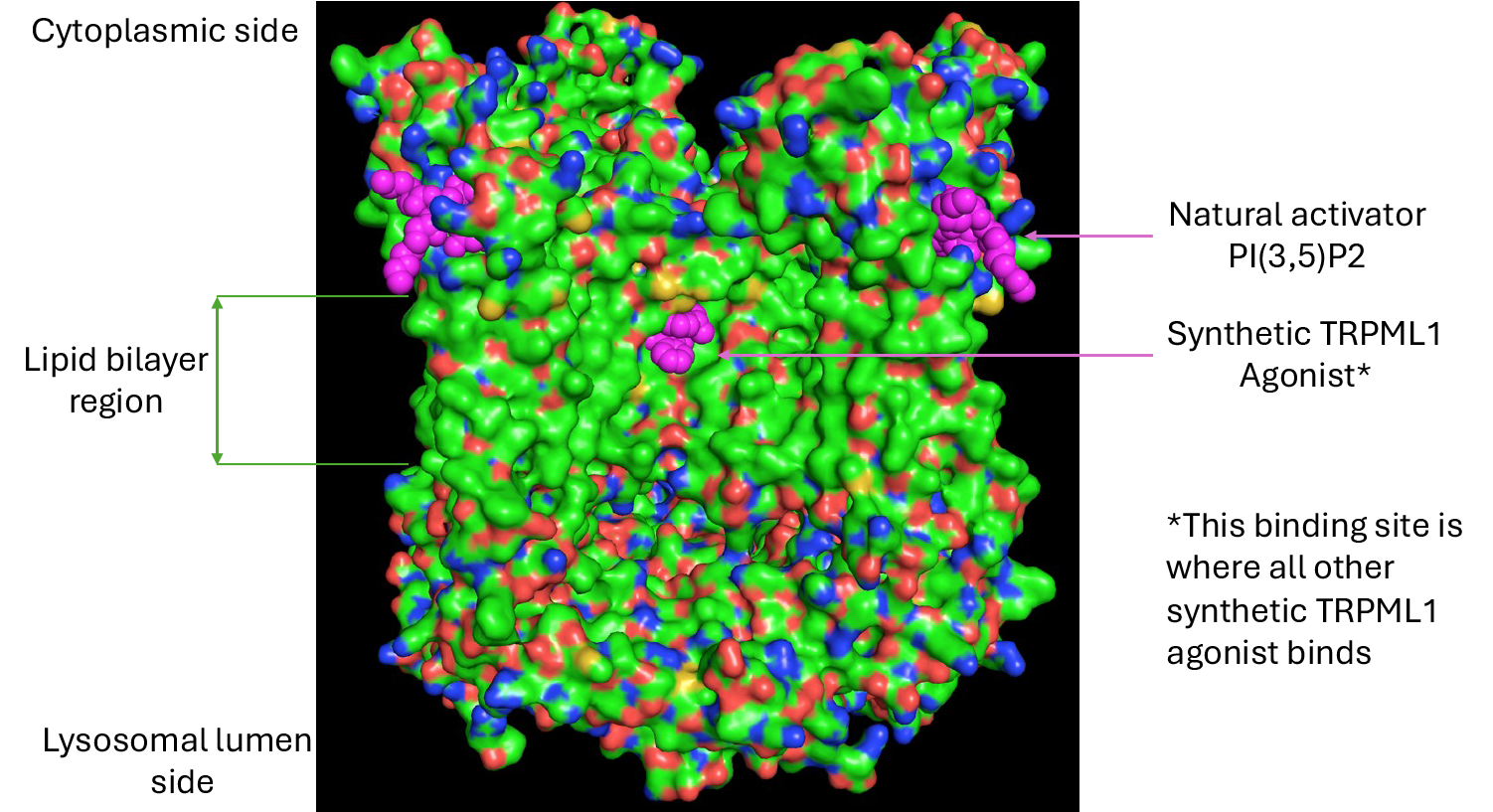
Figure 2.
High resolution Cryo-EM structure of TRPML1 with both natural activator PI(3,5)P2 and synthetic agonist bound to two different binding pockets, one is highly hydrophilic and the other is extremely hydrophobic and buried deep inside the protein and lipid bilayer.
Translational Research Challenges: Choosing the Right Models
A central challenge in translating TRPML1 agonists is identifying models that reflect the biology of aging. While many in vitro and in vivo models exist for AD and PD, few capture the age-related decline in autophagy–lysosomal pathway (ALP) function — the core defect TRPML1 agonism is designed to restore.
- Human iPSC-derived neurons (e.g., dopaminergic cells): These often show low TRPML1 and TFEB/TFE3 expression. In addition, culture stress can drive TFEB/TFE3 into the nucleus even without stimulation, masking TRPML1-dependent effects (unpublished Lysoway data). As such, they provide limited value for agonist testing.
- Genetic AD/PD mouse models: While useful for studying protein aggregation, they lack the aging component. Because the therapeutic rationale for TRPML1 focuses on reversing age-related ALP decline, aged models are more relevant.
Recent work underscores this. Culley et al. (2025) demonstrated that aged brains share features with early-stage AD and are acutely vulnerable to Aβ injury, while younger brains compensate. Consistent with this, we observed that progranulin, a key neuroprotective factor, is significantly reduced in aged versus young midbrains (unpublished Lysoway data).
Taken together, these findings highlight the goal of TRPML1 agonism: rejuvenating the brain’s intrinsic clearance machinery, which falters with age. For this reason, aging-based models, while imperfect, are essential for demonstrating in vivo efficacy and ensuring meaningful translation to the clinic.
Safety Considerations
TRPML1 is a ubiquitous lysosomal channel, expressed across virtually all tissues. Over- or prolonged activation therefore carries risks of exaggerated pharmacology and systemic toxicity. To maximize the therapeutic margin, several design principles are critical:
- High brain penetrance: Ensures therapeutic concentrations in the brain while minimizing peripheral exposure. At high doses, peripheral organ effects (e.g., in liver) have been observed in rodents and primates (internal and public reports). Agonists with lower brain-to-plasma ratios tend to have narrower margins.
- Balanced PK profiles: An excessively long systemic half-life may keep TRPML1 open too long, causing toxicity. Activation should be potent but temporally controlled. For example, in rats administered twice daily (BID) at high dose levels, tolerance dropped sharply, likely from prolonged activation (unpublished Lysoway data).
The ideal TRPML1 agonist for neurodegeneration is therefore one that combines high brain penetrance, oral bioavailability, potency, and carefully tuned PK — features we have prioritized in our candidate now entering clinical development.
Outlook and Clinical Considerations
The critical question now is whether TRPML1 agonists can deliver on their promise in patients. Early clinical trials will need to go beyond safety to capture pharmacodynamic signals — e.g. biomarkers of ALP restoration, lysosomal biogenesis, and aggregate clearance, as well as disease-related molecular changes. Such readouts in Phase 1b/2a will be vital to shaping Phase 2b studies designed to demonstrate true clinical benefit.
With growing momentum in the field and the progress achieved at Lysoway, the outlook is highly encouraging. For the first time, the possibility of rejuvenating the brain’s intrinsic clearance systems — rather than merely compensating after their failure — is within reach.
Conclusion
Antibody therapies have validated the concept of protein clearance but remain downstream, targeting consequences rather than root causes. TRPML1 agonists represent a fundamentally different approach: restoring the aging brain’s capacity to take care of itself.
At Lysoway Therapeutics, we have overcome the medicinal chemistry barriers that constrained the field and are now advancing a potentially first and best-in-class, brain-penetrant, orally bioavailable TRPML1 agonist into clinical development. If successful, this work could mark the beginning of a new era in treating AD, PD, and other age-related neurodegenerative diseases.
Contact Info
Contact us at Info@lysoway.com